2022_EBV
EBV Virus
-
Epstein-Barr virus는 헤르페스바이러스과의 바이러스 중 사람을 감염시키는 9가지 바이러스중에 하나이다. 사람에서 가장 흔이 발견되는 바이러스 중에 하나.
-
감염성 핵단구증이 가장 큰 원인이다. 림프구와 무관하게 위암, 비인두암을 일으키기도 하고, 인간면역결핍 바이러스와 함께 구강모백반증이나 중추신경계통 림프종을 일으키기도 한다. 매년 20만 이상의 암이 이 바이러스로 인해 발생한다.
-
침이나 생식기 분비액으로 전염될 수 있다.
-
대부분은 심각한 증상으로 발전하기 전에 후천 면역을 획득한다. 미국에서 조사한 결과, 성인들 중 90%가량이 이전에 감염된 흔적을 가지고 있는 것으로 나타났다. 많은 어린이들이 이 바이러스에 감염되지만 대부분 경미하고 짧은 증상만을 보인 뒤 정상으로 돌아간다. 청소년기에 감염될 경우 35~50%가 감염성 핵단구증을 앓는다.
chIP seq(Chromatin ImmunoPrecipitation followed by sequencing)
정의
NGS 기술을 활용한 세포내 전사조절인자 규명을 위해 이용된다. 특정 단백질의 세포내 전사 조절인자로서의 기능을 알고자 할때 특정 단백질이 binding하는 DNA 서열을 NGS로 sequencing하여 유전체 전체에서 binding 가능한 motif를 search한다.
원리
-
알고자 하는 특정 단백질을 in-vitro상에서 발현후 정제하여 해당 단백질에 특이적인 anti-body를 제작한다.
-
정제된 단백질과 whole genome을 binding 시킨 후 DNAase를 처리하여 단백질이 binding 되지 않은 비특이적인 DNA 서열은 모두 제거 되도록 한다.
-
정제된 단백질과 결합된 DNA 서열 겹합체를 앞서 제작한 anti-body를 이용해 분리해 낸다.
-
NGS sequencing을 수행한다.(TF binding site가 그리 길지 않은 점을 감안하여 1x 50 ~ 75bp로 sequencing한다.)
-
sequencing된 서열을 해당종의 genome서열에 mapping하여 consensus motif site를 확인 한다.
-
motif 서열을 기준으로 가장 가까운 유전자들을 대상으로 Functional annotation 수행으로 TFs의 downstream regulation study에 이용한다.
-
동일 condition에 해당하는 Input DNA(control)를 추가 실험 및 분석하여 peak calling시 bias를 제거 할 수 있다.
what is the plan?
- CTCF-binding site에서 human과 EBV virus의 DNA가 합쳐진 부분을 찾아서 어떤 chromosome에 binding이 많이 되어 사람에게 영향을 주는지 확인 해보는 것이 목표이다.
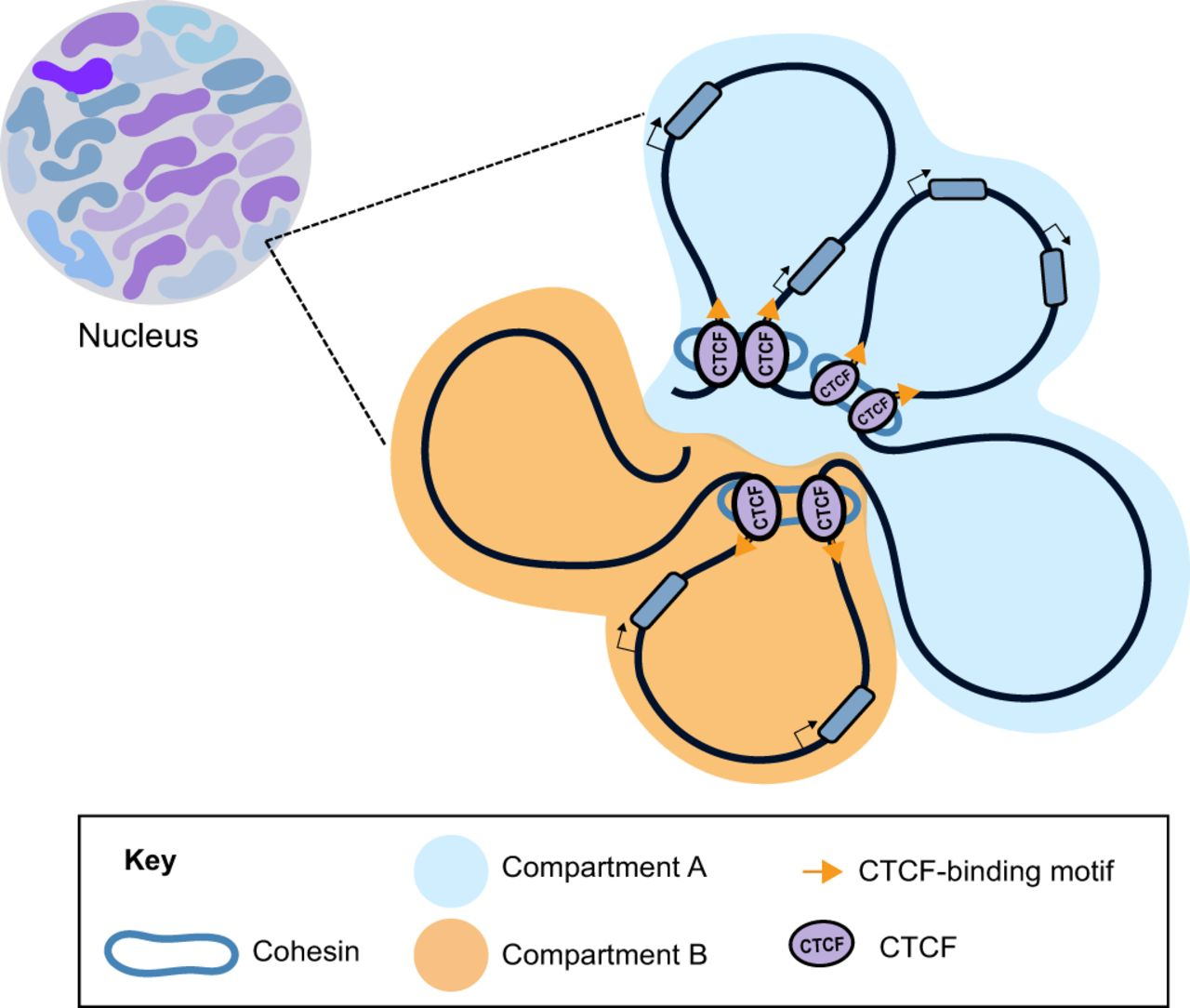
CTCF가 붙은 부분이 어디에 많이 분포되어있는지 보고싶달까..?
fastq파일 mapping
- NGS를 이용해서 만들어낸 fastq파일을 bwa를 통해서 mapping하는 과정을 진행했다.
bwa mem -t 20 -v 2 hg38_ebv_combined.fa CTCF-ab_1.trimmed.fastq CTCF-ab_2.trimmed.fastq
> CTCF-ab.bwa.samreference를 human과 EBV virus이 reference genome을 합친 새로운 .fa파일을 사용했다. 그 이유는 human과 EBV의 CTCF가 binding이 이루어지면 human안에 EBV의 서열의 일부가 삽입되는데 이것을 bwa를 통해 chimetic sequence의 정보를 정확하게 파악하기 위함이다. (다른 서열의 위치도 파악할 수 있다.)
A00718:399:H32CWDSX3:2:1101:13132:1219 147 chr6 36469195 60 66M35S = 36469186 -75
CAGTGGAAATTTGTACAGTGTAAACTGTGAAAGTGCTAAGTAGAGTGAACTTCAGAAATATTTTGAAGCTTTTGTTGCTGTTGTTGCTGGTGACATTTTAG
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
NM:i:0 MD:Z:66 M
C:Z:75M26S AS:i:66 XS:i:21 SA:Z:chr20,9662324,+,38M63S,60,0;sam파일안에 tag에 SA가 있다면 chimetic sequence라고 할 수 있는데, 또 다른 서열의 위치도 알 수 있다. 원래는 human:EBV로 구성된 sequence를 분석해볼려고 했는데, 처음은 일단 EBV:EBV인 sequence를 먼저 살펴보기로 했다. 왜냐하면 EBV의 sequence length가 사람에 비해서 엄청 짧기 때문에 IGV로 시각화 하기 더 편하다고 생각했기 때문이다. 그래서 mapping을 새로 해봤다.
bwa mem -t 20 -v 2 hg38.fa CTCF-ab_1.trimmed.fastq CTCF-ab_2.trimmed.fastq
> CTCF-ab.bwa.sam그리고 python package인 pysam을 이용해서 read들 중에 SA tag가 붙어있는 read만 출력시키도록 해봤다.
import pysam
samfile = pysam.AlignmentFile("CTCF-ab.bwa.sam", "rb")
for read in samfile.fetch():
for i in read.tags:
if i[0] == 'SA' and "NC.76005" in i[1]:
print(read)
break
samfile.close()A00718:399:H32CWDSX3:2:2678:10818:36260
TTTCGCGAGGTTAGGGACAACACGTTCCTCGACAATTTCGCGAGGTTAGGGACAACACGTTCCTCGACAAGTAC
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFMake new fastq file
이 read들을 cigar string에 맞게 read들을 쪼개서 새로운 fastq를 만들어보기로 했다.
import pysam
def sum(array):
count = 0
for i in range(len(array)):
count += int(array[i][1])
return count
token = "1:N:0:CTTGTA"
samfile = pysam.AlignmentFile("CTCF-ab.bwa.sam", "rb")
fastq = open("CTCF-ab.new.fastq", 'w')
for read in samfile.fetch():
for i in read.tags:
if i[0] == 'SA' and 'NC_007605' in i[1]:
count = 0
new = read.qname.split(':')
newString = '@'
for j in read.cigar:
# print(j)
if int(j[0]) == 0 or int(j[0]) == 4 or int(j[0]) == 5:
a= read.seq[count:(count + int(j[1]))]
b = "+\n" + read.qual[count:(count + int(j[1]))]
count += int(j[1])
for k in new:
newString += k + ':'
t = newString[:-1] + " " + token
if(a == ""):
pass
else:
result = t + '\n' + a + '\n' + b + '\n'
fastq.write(result)
newString = '@'
break
samfile.close()
fastq.close()새로운 fastq file
@A00718:399:H32CWDSX3:2:2678:10818:36260 1:N:0:CTTGTA
TTTCGCGAGGTTAGGGACAACACGTTCCTCGACAA
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
@A00718:399:H32CWDSX3:2:2678:10818:36260 1:N:0:CTTGTA
TTTCGCGAGGTTAGGGACAACACGTTCCTCGACAAGTAC
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF이 fastq파일을 다시 bwa를 통해 mapping해서 sam file로 만들었다. reference genome은 EBV virus의 genome을 사용했다.
그리고 이 sam파일을 read qname별로 새로운 배열을 만들어서 dataframe을 만들었다.
import pysam
import pandas as pd
def sum(read):
result = 0
for j in read.cigar:
result += j[1]
return result
url = ["2_CTCF.sa.bwa.sam", "CTCF-C-SNU719.sa.bwa.sam", "CTCF-ab_new.sa.bwa.sam", "/data3/projects/2022_KNU_EBV/aln/bwa/2021_SNU719/CTCF-ab_SA.bwa.sam"]
samfile = pysam.AlignmentFile("url[1], "rb")
array = []
tmp = []
name = []
count = 0
for read in samfile.fetch():
current = read.qname
if read.pos == -1:
pass
else:
a = read.pos
if current in name:
print("case1 " + current)
count += 1
if a in array[name.index(current)]:
pass
else:
array[name.index(current)].append(a)
else:
print("case2 " + current)
name.append(current)
array.append([])
array[name.index(current)].append(a)
newName = []
newArray = []
for i in range(0,len(name)):
print(name[i] + " : " + str(array[i]))
if len(array[i]) == 2:
newName.append(name[i])
newArray.append(array[i])
elif(len(array[i]) > 2):
newName.append(name[i])
newArray.append(array[i][0:2])
df = pd.DataFrame(newArray, index = newName)
df.to_csv('CTCF-C-SNU719.csv')
samfile.close() start end
A00125:510:HFN33DSX2:2:1107:31313:19492 7803 7640
A00125:510:HFN33DSX2:2:1122:15872:3677 7638 7483
A00125:510:HFN33DSX2:2:1135:23836:18067 7714 7478
A00125:510:HFN33DSX2:2:1153:30454:12493 8016 8033
A00125:510:HFN33DSX2:2:1237:13223:4476 24713 21641
... ... ...
A00125:510:HFN33DSX2:2:2501:26151:36667 7503 7899
A00125:510:HFN33DSX2:2:2528:18150:27258 7480 7637
A00125:510:HFN33DSX2:2:2543:9399:1705 7512 7928
A00125:510:HFN33DSX2:2:2570:1624:23719 39807 38493
A00125:510:HFN33DSX2:2:2575:17553:15608 7899 7634-
어느 region의 read가 어느 region으로 붙었는지 볼 수 있다.
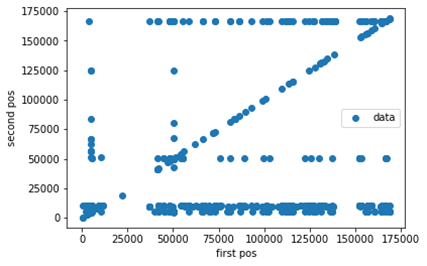
-
read들이 어떤 부분에 붙었는지 시각적으로 알 수 있다. (9739)
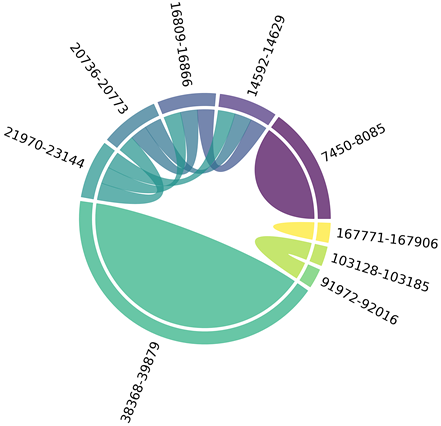
Human-EBV SA tag read 추출
하지만 EBV virus끼리 chimetic sequence를 만든 data는 뚜렷한 결과가 나오지 않았다. 그래서 human:EBV chimetic sequence를 추출해서 새로 mapping해보기로 했다.
애초에 이전에 했던 프로토콜은 모두 EBV안에 read들이 위치해있었기 때문에 시각화하기 편했는데 이번에는 한쪽에는 EBV position 한쪽에는 human position으로 시각화를 해야했기 때문에 어떻게 시각화를 할지 고민을 했다.
처음으로는 SA tag가 붙은 read들중에 human-EBV로 mapping되어있는 read를 따로 가져왔다.
token = "1:N:0:CTTGTA"
for read in samfile.fetch():
flag = 0
for i in read.tags:
ref = samfile.get_reference_name(read.tid)
if i[0] == 'SA':
if 'chr' in ref and 'NC_007605' in i[1]:
# human에 mapping되었고 그 다른부분이 EBV에 들어가 있는 경우
flag = 1
break
elif 'NC_007605' in ref and 'chr' in i[1]:
# EBV에 mapping되었고 그 다른부분이 human에 들어가 있는 경우
flag = 1
break
if flag == 1:
count = 0
new = read.qname
# print(new)
newString = '@' + new
print(read.cigar)
for j in read.cigar:
if int(j[0]) == 0 or int(j[0]) == 4 or int(j[0]) == 5:
# matching, SA, ZA가 붙은 경우
a = read.seq[count:(count + int(j[1]))]
b = "+\n" + read.qual[count:(count + int(j[1]))]
count += int(j[1])
name = newString + " " + token
# print(a)
# print(b)
if (a == ""):
pass
else:
result = name + '\n' + a + '\n' + b + '\n' # fastq file format에 맞게 만들어준다
print(result)
fastq.write(result)
newString = '@'+new
samfile.close()그리고 새로운 fastq file을 만들어서 human-ebv reference genome에 mapping을 했다.
bwa mem -t 20 -v 2 hg38_ebv_combined.fa CTCF-SNU719.trimmed.fastq CTCF-SNU719.trimmed.fastq
> CTCF-SNU719.bwa.sammapping 정보들이 chromosome별로, position별로 다 다르게 mapping이 되어있었기 때문에 일단 먼저 bedtools를 이용해서 특정 구간에서 mapping되어있는지 기준을 통해 merge했다.
bedtools merge -i CTCF-SNU719.bwa.sam -c 1 -o collapse > CTCF-SNU719.bedread들이 많은 특정구간을 구해서 거기 안에 속해있는 read들이 어떤 것들이 있는지 annotation이 되어있다.
#bed file format
NC_007605.1 2091 2164 A00718:399:H32CWDSX3:2:1251:8241:31610,
A00718:399:H32CWDSX3:2:1636:12075:10222이 bed file을 통해서 EBV의 어떤 유전자가 human genome에 가장 많이 붙는지를 알아보고 싶었기 때문에 구간별로 read count를 세고 그 구간에 존재하는 유전자의 정보를 확인해보기로 했다.
일단 bed file 자체는 chromosome별로 정리가 되어있지 않았기 때문에 chr별로 발현량을 체크하기 위해 파일을 분리했다.
bed = open("/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/CTCF-C-SNU719.bed")
url = "/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/SNU719_chr/"
chromosome = ['chr1']
prev = "chr1"
temparray = ""
array = ""
while True:
line = bed.readline().split()
if not line:
urls = url + prev + ".bed"
urlTemp = open(urls, "w")
urlTemp.write(str(temparray))
urlTemp.close()
break
newtmp = ""
chrtmp = line[0].split(',')[0]
starttmp = line[1].split(',')[0]
endtmp = line[2].split(',')[0]
qnametmp = line[3].split(',')
for i in range(0,len(qnametmp)):
newtmp += qnametmp[i]
newtmp += ','
array = chrtmp + '\t' +starttmp +'\t' + endtmp + '\t' + newtmp[0:-1]
if chrtmp not in chromosome:
chromosome.append(chrtmp)
urls = url + prev+ ".bed"
urlTemp = open(urls ,"w")
urlTemp.write(str(temparray))
urlTemp.close()
temparray = ""
temparray+=array + '\n'
prev = chrtmp
else:
temparray+=array + '\n'
bed.close()그리고 chromosome별로 시각화를 진행해보았다.
- EBV - Human chimetic sequence chromosome 20 gene expression
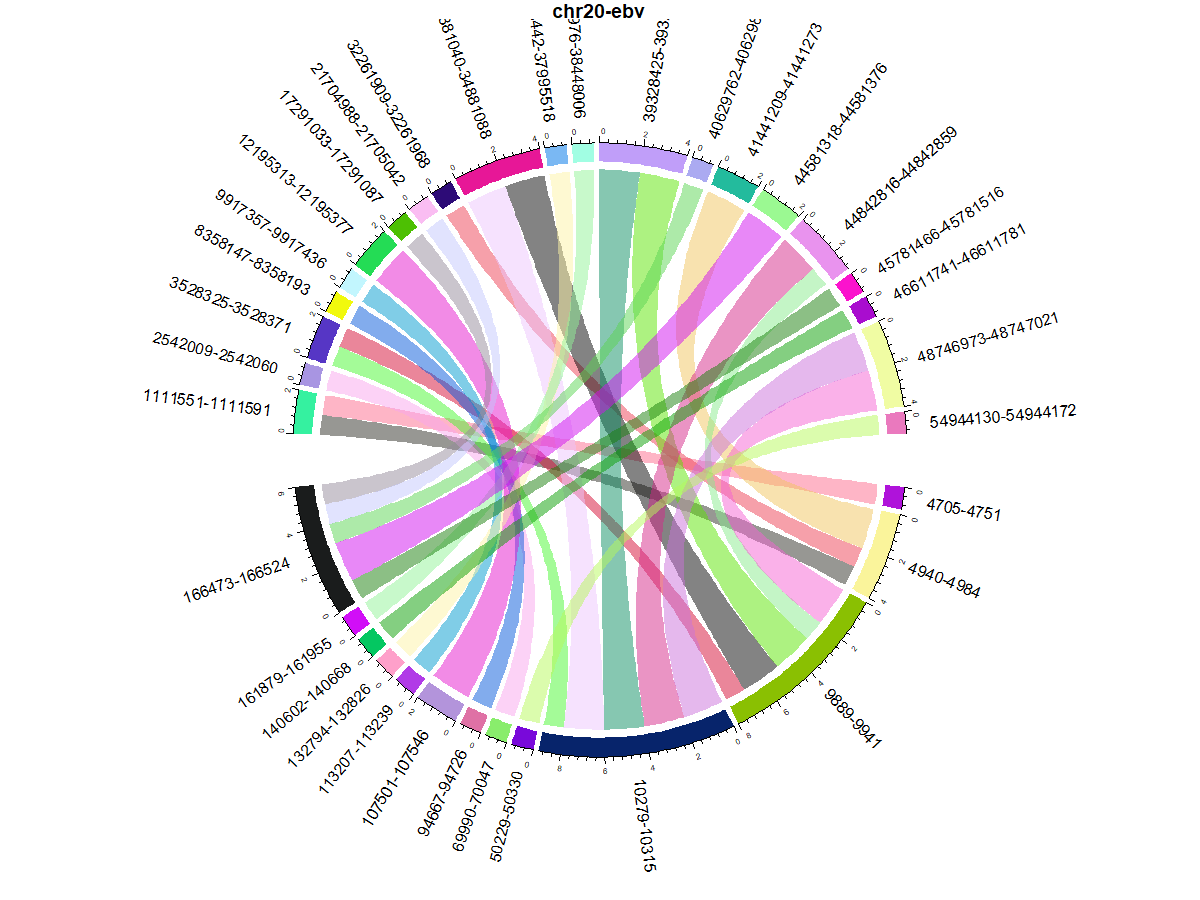
Sector 별 발현량 count
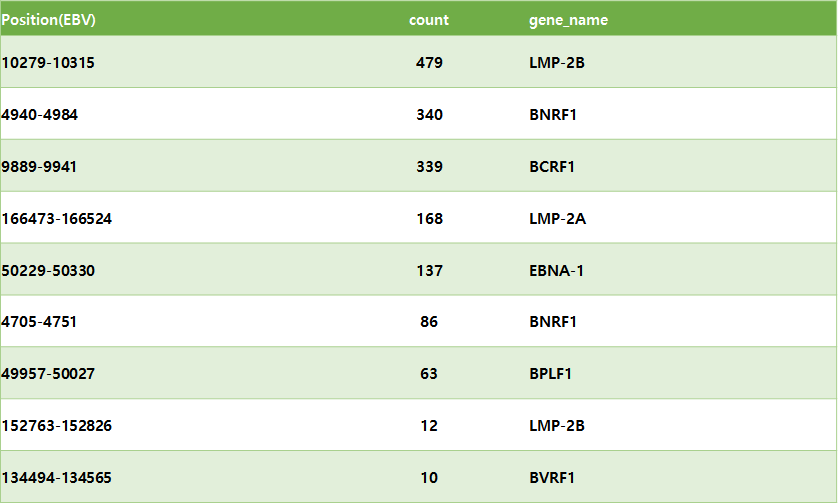
chr1부터 chrY까지 시각화를 해보았는데 가장 눈에 띄는 것은 “10279-10315”와 같은 EBV virus의 특정 부분이 많이 보였다는 것이다. 그래서 어떤 sector가 많이 발현됐고 그 sector에는 어떤 유전자가 있는지 확인해보기로 했다.
import pandas as pd
total = []
for i in range(1,23):
total.append(str(i))
total.append("X")
total.append("Y")
array = []
name_array = []
for k in total:
csv_test = pd.read_csv("/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/chr_bed/chr" + k +".csv", index_col = 0)
for i in range(0,len(csv_test.index)):
tmp = csv_test.iloc[i].sum()
name = csv_test.iloc[i].name
if tmp==0:
pass
else:
if name not in name_array:
name_array.append(name)
array.append(tmp)
else:
array[name_array.index(name)] += tmp
df = pd.DataFrame(array,columns = ['count'], index = name_array)
df_sorted = df.sort_values(by = df.columns[0],ascending=False)
print(df_sorted)
df_sorted.to_csv("/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/SNU719_count/total_count.csv")여기에 어떤 유전자가 있는지 확인해보기 위해 htseq package를 통해서 gtf파일을 불러온 다음에 유전자 위치에 대한 정보를 parsing했다.
import HTSeq
import pandas as pd
name = []
pos = []
ebv = "/data3/PUBLIC_DATA/ref_genomes/Human_gammaherpesvirus_4_EBV/NC_007605.1.gtf"
human = "/data3/PUBLIC_DATA/ref_genomes/homo_sapiens/hg38/hg38.gtf"
def gtf_count():
gtf_file = HTSeq.GFF_Reader(ebv)
print(gtf_file)
exons = HTSeq.GenomicArrayOfSets("auto", stranded=True)
for feature in gtf_file:
exons[feature.iv] += feature.attr["transcript_id"]
# print(feature.attr["transcript_id"])
array = feature.get_gff_line().split('\t')
if "gene_name" in feature.attr:
name.append(feature.attr["gene_name"])
else:
name.append(feature.attr["transcript_id"])
pos.append([int(array[3]),int(array[4])])
df = pd.DataFrame(pos, index = name, columns=["start","end"])
print(df)
df.to_csv("/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/SNU719_count/ebvPos.csv")
gtf_count()이 두 코드를 통해서 만들어진 파일들로 구간에 존재하는 유전자를 매칭했다.
import pandas as pd
ebvannot = pd.read_csv('/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/SNU719_count/ebvPos.csv',index_col=0)
chrcount = pd.read_csv('/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/SNU719_count/total_count.csv',index_col=0)
array = [[] * 18 for _ in range(18)]
other = 'LMP-2B'
for i in range(1,len(ebvannot.index)):
# print(ebvannot.index[i],ebvannot['start'][i],ebvannot['end'][i])
tmp = [int(ebvannot['start'][i]),int(ebvannot['end'][i]), ebvannot.index[i]]
index = int(ebvannot['start'][i]) // 10000
array[index].append(tmp)
annotarray = []
for k in chrcount.index:
flag = 0
temp = list(map(int,k.split('-')))
pos = temp[0] // 10000
for i in array[pos]:
if(i[0] <= temp[0] and temp[1] <= i[1]):
print("yes : " + i[2])
annotarray.append(i[2])
flag = 1
break
if flag == 0:
print("no : " + other)
annotarray.append(other)
# print(temp)
df = pd.DataFrame(annotarray, index = chrcount.index, columns=['gene_name'])
new_df =pd.concat([chrcount,df], axis=1)
print(new_df)
new_df.to_csv("/data3/projects/2022_KNU_EBV/trimmed/2021_SNU719/yj/SNU719_count/total_count_annot.csv")gtf를 담은 파일의 데이터 수가 엄청 많았기 때문에 hashing을 이용해서 구간을 나누어서 찾을 수 있도록 했다.
Motif 확인
Human의 유전자도 확인해보고 싶었는데 너무 scatter하게 퍼져있어서 결과가 깔끔하게 나오지 않았다. 코드로 작업해서 만들기에는 gtf파일의 크기가 너무 커서 탐색을 하기에 time complexity가 너무 컸기 때문에 count수가 높은 read들의 position을 확인해봤는데, exon이 아닌 intron이 대부분이었다..
뚜렷한 특징이 잘 안보였기 때문에 마지막으로 chr마다 sequence의 특정 motif가 있는지 확인해보기로 했다.
-
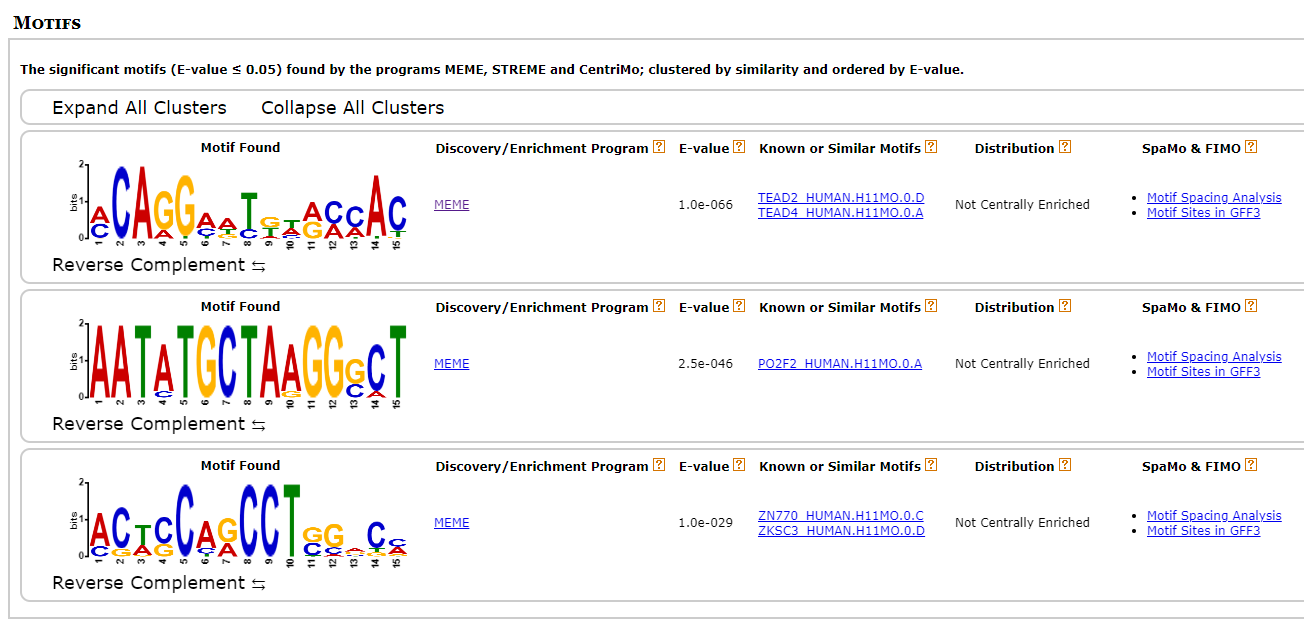
4940
-
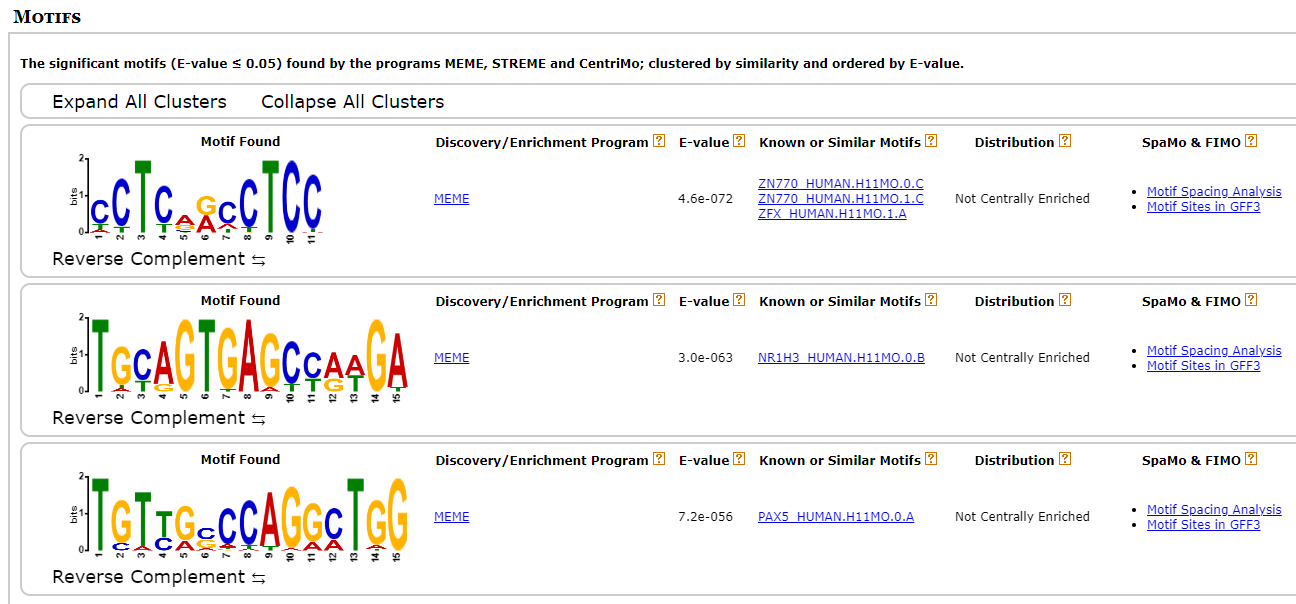
9889
-
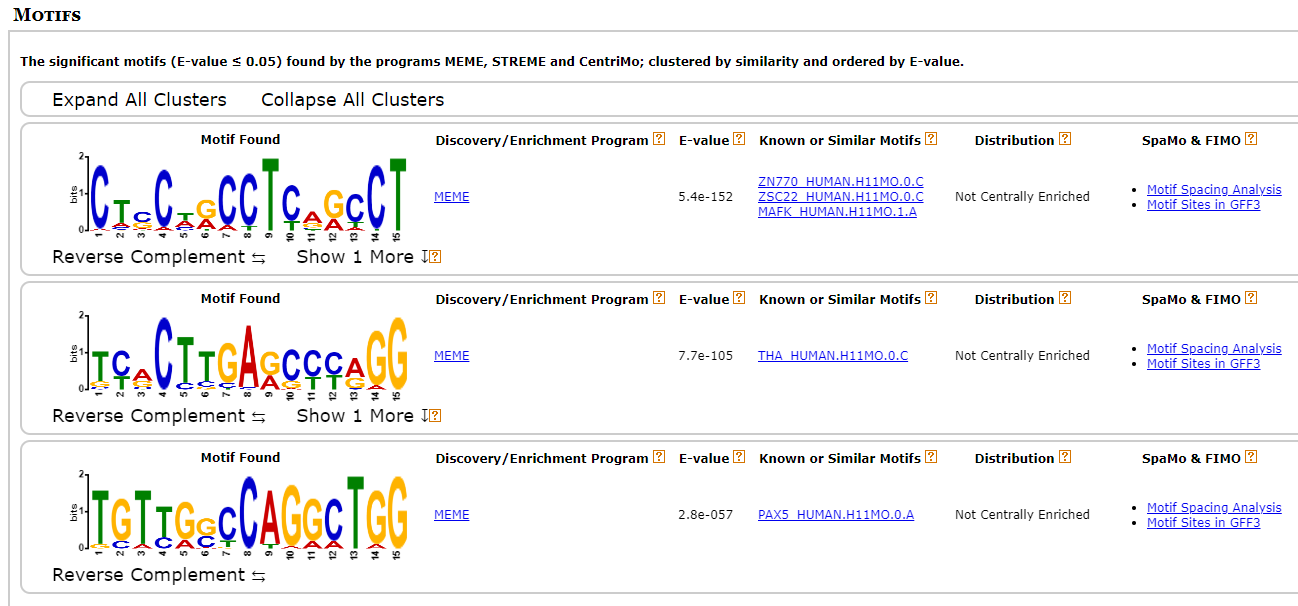
10279
-
all(4940 + 9889 + 10279)
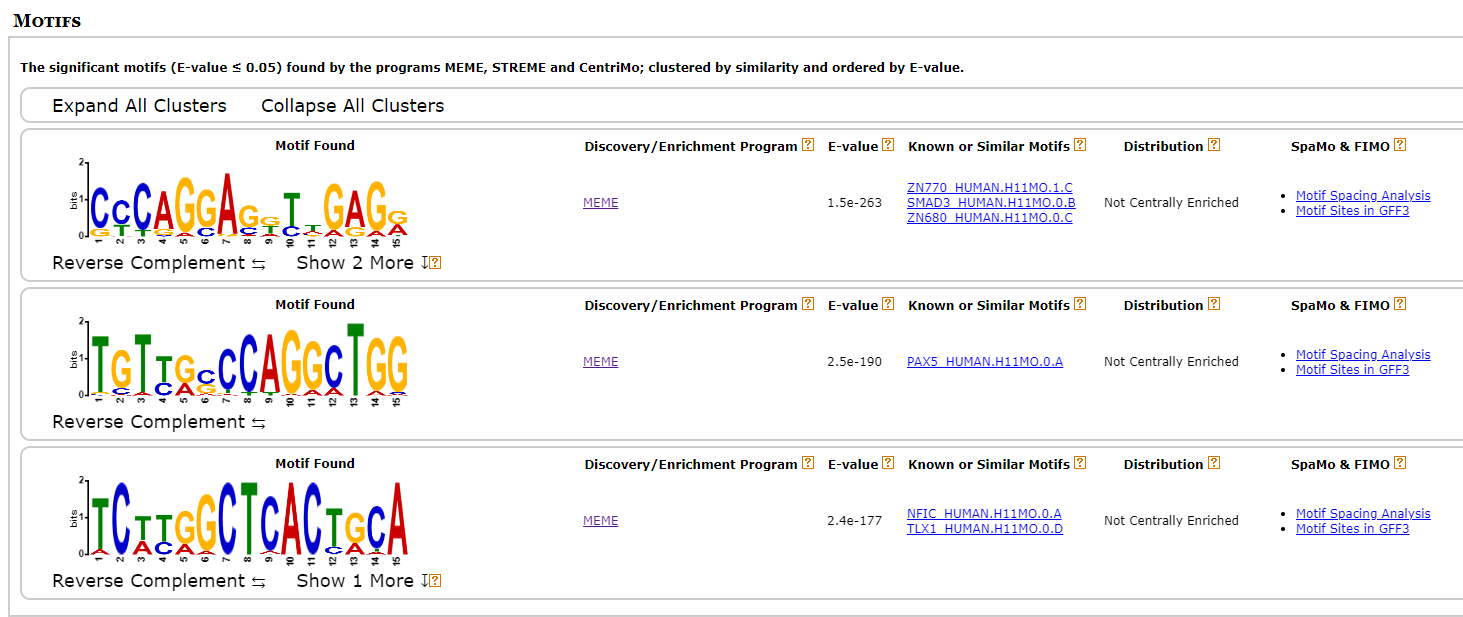
-
other

확실히 binding이 많이 되는 site와 아닌 site의 motif가 다른 것 같다. 일단 세 point에서 가장 많이 나온 “ZN770”과 “PAX5” 유전자를 살펴보기로 했다.
ZN770은 DNA polymerase2에 특이적으로 결합하는 motif인데 딱히 눈에 띄는 유전자는 아니었다. 반면에 PAX5는 달랐다.
PAX5는 EBNA1이라는 ebv 유전자의 발현 개수를 조절해주는 인자인데 이 EBNA1이 암과 증상을 유발하는 key motif 였던 것이다. 위치적으로도 그렇고 EBNA1은 11400 정도 되는 pos를 가지고 있었는데 이 PAX5가 나온 부분은 공교롭게도 1000bps정도밖에 차이가 나지 않았다.
